FDA, EMA: Новые правила ИИ в фармразработке с 2026 года

Pharma AI R&D faces new regulatory hurdles in 2026 as FDA & EMA define credible AI evidence & lifecycle audits. Prepare now!
2026: FDA и EMA устанавливают правила для ИИ в разработке лекарств
С 2026 года FDA и EMA вводят новые регуляторные требования к ИИ в фармразработке, трансформируя свою роль из наблюдателей в строгих контролёров R&D-процессов. Эти правила обязывают фармацевтические компании соблюдать жёсткие протоколы валидации ИИ-моделей, управления рисками и документирования их жизненного цикла. Компании должны предоставлять исчерпывающие доказательства применения ИИ, вести полный учёт, оценивать риски и подтверждать эффективность и безопасность своих моделей.
Надзорные органы теперь требуют полной документации, чёткого человеческого контроля и надёжного управления данными, исключая любую терпимость к моделям типа «чёрный ящик». Хотя эти требования могут увеличить начальные сроки проектов, они гарантируют, что в создании новых лекарств будут использоваться только безопасные и тщательно проверенные ИИ-модели.
Какие новые регуляторные требования к ИИ в фармразработке действуют с 2026?
С 2026 года FDA и EMA требуют, чтобы применение ИИ в разработке лекарств соответствовало чётко определённой структуре. Эта структура включает ясный контекст использования (COU), тщательную оценку рисков, надёжный план валидации, строгое управление данными, полную документацию модели, формализованные SOP для контроля изменений и непрерывный мониторинг жизненного цикла. Отсутствие ключевых компонентов, особенно планов контроля изменений, теперь приводит к серьёзным задержкам при регуляторном рассмотрении.
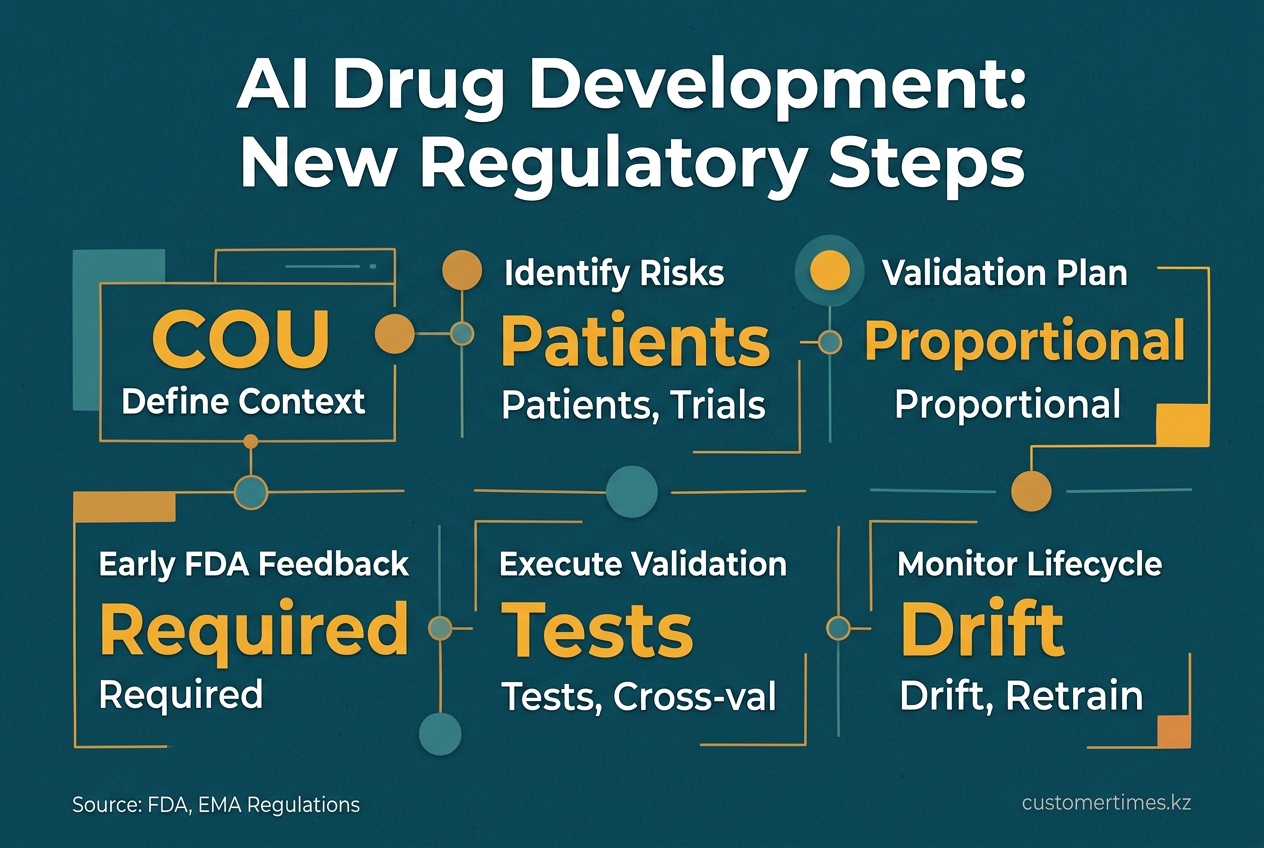
Черновик правил 2025 года - семь шагов, один контекст
Новые правила требуют от разработчиков чётко определить контекст использования (COU) ИИ-модели и связанные с ним риски. На этой основе создаётся соразмерный план валидации, документируются все методологии и обеспечивается управление жизненным циклом модели, включая контроль версий и получение ранних отзывов от регуляторов.
Согласно новым правилам, спонсоры должны определить конкретный контекст использования (COU) для ИИ-модели и решение, на которое она влияет. На основе этого они обязаны:
- Выявить все риски для пациентов, целостности испытаний и качества продукта.
- Создать план валидации, соразмерный выявленным рискам.
- Получить ранний отзыв FDA по плану валидации.
- Выполнить план валидации (статистические тесты, стресс-тесты, кросс-валидация).
- Архивировать все методологии, исходные данные, результаты и отклонения.
- Определить и задокументировать все методологии, исходные данные и результаты.
- Мониторить модель после внедрения для отслеживания дрейфа и переобучать её при строгом контроле изменений.
Для приложений с высоким риском (например, подбор дозы, ключевые конечные точки или выпуск продукции) FDA теперь ожидает предварительных встреч и раскрытия алгоритмов с детализацией, эквивалентной традиционной документации CMC.
«Спонсорам следует встретиться с агентством перед использованием ИИ-модели высокого риска в критическом решении.»
- Проект руководства FDA, январь 2025
Рецензенты агентств применяют эту структуру с 2016 года к более чем 500 заявкам, преимущественно в онкологии и неврологии.
Совместные принципы 2026 - десять заповедей для полного жизненного цикла
В январе 2026 года FDA и EMA опубликовали совместные «Руководящие принципы надлежащей практики ИИ в разработке лекарств», согласовав свои ожидания. Несмотря на рекомендательный характер, эти десять принципов формируют основу будущих обязательных требований:
- Человеко-ориентированный дизайн с чёткими возможностями для вмешательства человека.
- Стандарты доказательности, основанные на оценке рисков, а не универсальный подход.
- Соблюдение установленных стандартов (ISO 13485, GAMP5, ICH Q9).
- Чёткий COU и соответствующая команда экспертов.
- Надёжное управление данными, включая отслеживание происхождения и контроль версий.
- Тщательная документация по разработке модели и полный аудиторский след.
- Метрики производительности, напрямую связанные с клинической значимостью.
- Комплексное управление жизненным циклом с протоколами обнаружения дрейфа.
- Прозрачная коммуникация между спонсором и регуляторами.
Подписанные также EMA, эти принципы напрямую связывают валидацию ИИ с требованиями целостности данных Приложения 11 ЕС и инспекциями GxP. Это означает, что аргументы о «чёрном ящике» не выдержат проверок на месте.
Как теперь выглядит пакет документов для подачи
Хотя регуляторы избегают жёстких шаблонов, идеальный пакет доказательств для ИИ-модели должен содержать следующие компоненты. Элементы, отмеченные как критические, часто запрашиваются дополнительно, если отсутствуют в первоначальной подаче.
| Компонент | Типичные артефакты | Риск при проверке, если отсутствует |
|---|---|---|
| Заявление о COU | Предполагаемое решение, критерии успеха | Серьёзный |
| Досье по управлению данными | Исходные таблицы, скрипты очистки, метрики QC | Критический |
| Спецификация модели | Диаграмма архитектуры, гиперпараметры, логи обучения | Критический |
| Отчёт о валидации | Внутренние/внешние выборки, ROC/AUC, доверительные интервалы | Серьёзный |
| Модуль объяснимости | Графики SHAP/LIME, извлечение правил, сопоставление с экспертами | Серьёзный |
| SOP контроля изменений | Хеширование версий, триггеры переобучения, план отката | Критический |
| План постмаркетинга | Дашборд производительности, периодичность обновлений, лимиты сигналов | Серьёзный |
Анализ ранних заявок показывает, что отсутствие SOP по контролю изменений с высокой вероятностью приведёт к остановке 90-дневного цикла рассмотрения, даже при высокой прогностической точности модели.
Разрыв скорости - почему ИИ обгоняет лабораторию
Несмотря на ужесточение требований к валидации, ИИ-модели генерируют тысячи потенциальных кандидатов в лекарства еженедельно, создавая так называемый «разрыв скорости»: ИИ предлагает молекулы быстрее, чем GxP-лаборатории могут их синтезировать и протестировать. Это приводит к значительным операционным задержкам. Для преодоления разрыва спонсоры внедряют гибридный подход: сначала прогнозы ИИ фильтруются через QSP- или PBPK-симуляции, и только затем начинается работа в лаборатории. Такой подход, по отзывам, сокращает число неудач на поздних стадиях на 23%, оставаясь в рамках новых правил.
От пилотов к платформам - что меняют спонсоры в 2026
- Изолированные PoC-команды интегрируются в межфункциональные комитеты по ИИ, включающие экспертов по регуляторике, QA, биостатистике и IT-безопасности.
- Для пилотных проектов с генеративным ИИ теперь требуется задокументированный COU ещё до утверждения бюджета. Исследование MIT 2025 года показало, что 95% пилотов, пропускавших этот шаг, потерпели неудачу.
- В контракты с CRO всё чаще включают пункты о соответствии данных принципам FAIR, чтобы предотвратить возражения регуляторов против непрозрачных аутсорсинговых моделей.
- Вендоры изначально встраивают в свои платформы API для аудиторского следа, чтобы избежать дорогостоящих доработок после запроса агентства.
«Регуляторная ясность - одно из трёх главных препятствий для более широкого внедрения ИИ в фарме.»
- Опрос индустрии, процитированный в анализе трендов 2026
Влияние на стоимость и сроки - первые метрики
Вопреки опасениям о задержках, компании, первыми внедрившие новые практики, сообщают о сокращении общего времени рассмотрения заявок благодаря заблаговременной подготовке документации. Исследование 42 заявок в FDA за 2025 год показало, что заявители, проводившие предварительные встречи с агентством, сократили сроки рассмотрения в среднем на 92 дня по сравнению с теми, кто этого не делал. Эта выгода перевешивает затраты времени на внутреннюю подготовку.
Глобальная рябь - размышления EMA и импульс ICH
Размышлятельный документ EMA (сентябрь 2024) уже подчёркивал важность прозрачности, валидации и человеческого контроля. Совместные принципы 2026 года ускоряют глобальную гармонизацию и, как ожидается, лягут в основу будущего руководства ICH Q-AI. Это руководство призвано унифицировать терминологию валидации ИИ для регуляторов по всему миру, включая PMDA (Япония) и Health Canada. В результате спонсоры, планирующие международные исследования, уже на старте разрабатывают протоколы, совместимые с требованиями и FDA, и EMA.
Что будет дальше
Регуляторы дали понять, что формальные руководства превратят эти принципы в обязательные нормы в течение 18 - 24 месяцев. Ожидается, что эти документы введут многоуровневую систему соответствия: меньшая регуляторная нагрузка для ИИ на этапе исследований и полная GMP-валидация для ИИ, влияющего на спецификации продукта или выпуск партий. До тех пор агентства советуют спонсорам документировать всю деятельность с ИИ так, как если бы правила уже действовали, поскольку любая модель, использованная в заявке после 2026 года, будет оцениваться по новой структуре.
Компании, игнорирующие валидацию ИИ, рискуют повторить судьбу тех, кто медлил с переходом на электронные подачи: получить отказ в приёме документов или, что хуже, столкнуться с задержками вывода на рынок, пока конкуренты с готовым к аудиту ИИ выходят вперёд.